Creutzfeldt-Jakobs sygdom
Creutzfeldt-Jakobs sygdom (CJD) er en form for hjerneskade, der fører til et hurtigt fald i bevægelse og tab af mental funktion.
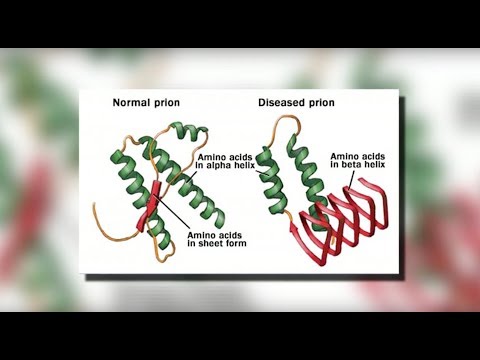
CJD er forårsaget af et protein kaldet en prion. En prion får normale proteiner til at folde sig unormalt. Dette påvirker andre proteins evne til at fungere.
CJD er meget sjælden. Der er flere typer. De klassiske typer CJD er:
- Sporadic CJD udgør de fleste sager. Det forekommer uden kendt grund. Den gennemsnitlige alder, hvor den starter, er 65 år.
- Familiel CJD opstår, når en person arver den unormale prion fra en forælder (denne form for CJD er sjælden).
- Erhvervet CJD inkluderer variant CJD (vCJD), formen relateret til gal ko sygdom. Iatrogen CJD er også en erhvervet form for sygdommen. Iatrogen CJD ledes undertiden gennem en blodprodukttransfusion, transplantation eller kontaminerede kirurgiske instrumenter.
Variant CJD er forårsaget af at spise inficeret kød. Infektionen, der forårsager sygdommen hos køer, menes at være den samme, der forårsager vCJD hos mennesker.
Variant CJD forårsager mindre end 1 procent af alle CJD-tilfælde. Det har tendens til at påvirke yngre mennesker. Færre end 200 mennesker verden over har haft denne sygdom. Næsten alle sager opstod i England og Frankrig.
CJD kan være relateret til flere andre sygdomme forårsaget af prioner, herunder:
- Kronisk spildesygdom (findes i hjorte)
- Kuru (berørte hovedsagelig kvinder i New Guinea, der spiste hjernen hos døde slægtninge som en del af et begravelsesritual)
- Scrapie (findes hos får)
- Andre meget sjældne arvelige sygdomme hos mennesker, såsom Gerstmann-Straussler-Scheinker sygdom og dødelig familiær søvnløshed
CJD-symptomer kan omfatte et af følgende:
- Demens, der hurtigt bliver værre over et par uger eller måneder
- Sløret syn (nogle gange)
- Ændringer i gang (gå)
- Forvirring, desorientering
- Hallucinationer (ser eller hører ting der ikke er der)
- Manglende koordination (for eksempel snuble og falde)
- Muskelstivhed, trækninger
- Føler nervøs, hoppende
- Personlighedsændringer
- Søvnighed
- Pludselige rykkende bevægelser eller anfald
- Problemer med at tale
Tidligt i sygdommen viser nervesystemet og mental undersøgelse problemer med hukommelse og tænkning. Senere i sygdommen kan en motorisk systemundersøgelse (en undersøgelse for at teste muskelreflekser, styrke, koordination og andre fysiske funktioner) vise:
- Unormale reflekser eller øgede normale refleksresponser
- Forøgelse i muskeltonus
- Muskeltrækninger og spasmer
- Stærkt skræmmende svar
- Svaghed og tab af muskelvæv (spild af muskler)
Der er tab af koordination og ændringer i lillehjernen. Dette er det område af hjernen, der styrer koordinationen.
En øjenundersøgelse viser områder med blindhed, som personen muligvis ikke bemærker.
Test, der bruges til at diagnosticere denne tilstand, kan omfatte:
- Blodprøver for at udelukke andre former for demens og søge efter markører, der undertiden opstår med sygdommen
- CT-scanning af hjernen
- Elektroencefalogram (EEG)
- MR i hjernen
- Spinal tap for at teste for et protein kaldet 14-3-3
Sygdommen kan kun bekræftes med hjernebiopsi eller obduktion. I dag er det meget sjældent, at der udføres en hjernebiopsi for at lede efter denne sygdom.
Der er ingen kendt kur mod denne tilstand. Forskellige lægemidler er blevet forsøgt at bremse sygdommen. Disse inkluderer antibiotika, medicin mod epilepsi, blodfortyndende midler, antidepressiva og interferon. Men ingen fungerer godt.
Målet med behandlingen er at give et sikkert miljø, kontrollere aggressiv eller ophidset adfærd og imødekomme personens behov. Dette kan kræve overvågning og hjælp i hjemmet eller i en plejefacilitet. Familierådgivning kan hjælpe familien med at klare de ændringer, der er nødvendige for hjemmeplejen.
Mennesker med denne tilstand kan have brug for hjælp til at kontrollere uacceptabel eller farlig adfærd. Dette indebærer belønning af positiv adfærd og ignorering af negativ adfærd (når det er sikkert). De har muligvis også brug for hjælp til at orientere sig i deres omgivelser. Nogle gange er der brug for medicin for at hjælpe med at kontrollere aggression.
Personer med CJD og deres familie skal muligvis søge juridisk rådgivning tidligt i løbet af lidelsen. Advance-direktiv, fuldmagt og andre juridiske handlinger kan gøre det lettere at træffe beslutninger om pleje af den person med CJD.
Resultatet af CJD er meget dårligt. Mennesker med sporadisk CJD er ude af stand til at passe sig selv inden for 6 måneder eller mindre efter, at symptomerne begynder.
Forstyrrelsen er dødelig på kort tid, normalt inden for 8 måneder. Folk, der har variant CJD, bliver værre langsommere, men tilstanden er stadig dødelig. Et par mennesker overlever så længe som 1 eller 2 år. Dødsårsagen er normalt infektion, hjertesvigt eller åndedrætssvigt.
Forløbet af CJD er:
- Infektion med sygdommen
- Alvorlig underernæring
- Demens i nogle tilfælde
- Tab af evne til at interagere med andre
- Tab af evnen til at fungere eller passe sig selv
- Død
CJD er ikke en medicinsk nødsituation. At blive diagnosticeret og behandlet tidligt kan dog gøre symptomerne nemmere at kontrollere, give patienterne tid til at udarbejde forhåndsdirektiver og forberede sig på livets afslutning og give familier ekstra tid til at ordne sig med tilstanden.
Medicinsk udstyr, der kan være forurenet, skal tages ud af drift og bortskaffes. Folk, der vides at have CJD, bør ikke donere en hornhinde eller andet kropsvæv.
De fleste lande har nu strenge retningslinjer for håndtering af inficerede køer for at undgå overførsel af CJD til mennesker.
Overførbar spongiform encefalopati; vCJD; CJD; Jacob-Creutzfeldt sygdom
 Creutzfeldt-Jakobs sygdom
Creutzfeldt-Jakobs sygdom Centralnervesystemet og det perifere nervesystem
Centralnervesystemet og det perifere nervesystem
Bosque PJ, Tyler KL. Prioner og prionsygdomme i centralnervesystemet (overførbare neurodegenerative sygdomme). I: Bennett JE, Dolin R, Blaser MJ, red. Mandell, Douglas og Bennett's principper og praksis for smitsomme sygdomme. 9. udgave Philadelphia, PA: Elsevier; 2020: kap. 179.
Geschwind MD. Prion sygdomme. I: Daroff RB, Jankovic J, Mazziotta JC, Pomeroy SL, red. Bradleys neurologi i klinisk praksis. 7. udgave Philadelphia, PA: Elsevier; 2016: kap 94.
Interessante Publikationer.

Forskellen mellem lupus og RA
