Cystisk fibrose

Cystisk fibrose er en sygdom, der får tykt, klæbrig slim til at opbygges i lungerne, fordøjelseskanalen og andre områder af kroppen. Det er en af de mest almindelige kroniske lungesygdomme hos børn og unge voksne. Det er en livstruende lidelse.
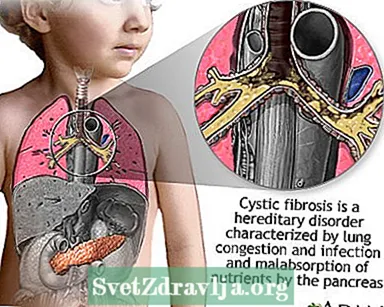
Cystisk fibrose (CF) er en sygdom, der overføres gennem familier. Det er forårsaget af et defekt gen, der får kroppen til at producere unormalt tyk og klæbrig væske, kaldet slim. Dette slim bygger sig op i lungerne og i bugspytkirtlen.
Opbygningen af slim resulterer i livstruende lungeinfektioner og alvorlige fordøjelsesproblemer. Sygdommen kan også påvirke svedkirtlerne og en mands reproduktionssystem.
Mange mennesker bærer et CF-gen, men har ikke symptomer. Dette skyldes, at en person med CF skal arve 2 defekte gener, 1 fra hver forælder. Nogle amerikanere har CF-genet. Det er mere almindeligt blandt dem af nordlig eller centraleuropæisk afstamning.
De fleste børn med CF er diagnosticeret i alderen 2, især da screening af nyfødte udføres over hele USA. For et lille antal opdages sygdommen først i en alder af 18 år. Disse børn har ofte en mildere form for sygdommen.
Symptomer hos nyfødte kan omfatte:
- Forsinket vækst
- Manglende vægt i normal i barndommen
- Ingen afføring i de første 24 til 48 timer i livet
- Salt med smagende hud
Symptomer relateret til tarmfunktion kan omfatte:
- Mavesmerter fra svær forstoppelse
- Øget gas, oppustethed eller en mave, der ser hævet ud (udspilet)
- Kvalme og tab af appetit
- Afføring, der er bleg eller lerfarvet, ildelugtende, har slim eller flyder
- Vægttab
Symptomer relateret til lunger og bihuler kan omfatte:
- Hoste eller øget slim i bihulerne eller lungerne
- Træthed
- Næseoverbelastning forårsaget af næsepolypper
- Gentagne episoder af lungebetændelse (symptomer på lungebetændelse hos en person med cystisk fibrose inkluderer feber, øget hoste og åndenød, øget slim og appetitløshed)
- Sinus smerter eller tryk forårsaget af infektion eller polypper
Symptomer der kan bemærkes senere i livet:
- Infertilitet (hos mænd)
- Gentagen betændelse i bugspytkirtlen (pancreatitis)
- Åndedrætssymptomer
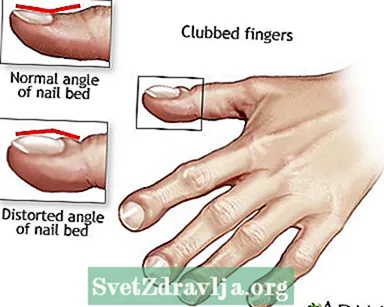
- Knebede fingre
En blodprøve udføres for at hjælpe med at opdage CF. Testen ser efter ændringer i CF-genet. Andre tests, der bruges til at diagnosticere CF, inkluderer:
- Immunoreaktivt trypsinogen (IRT) test er en standard nyfødt screening test for CF. Et højt niveau af IRT antyder mulig CF og kræver yderligere test.
- Sweatchlorid test er standard diagnostisk test for CF. Et højt saltniveau i personens sved er et tegn på sygdommen.
Andre tests, der identificerer problemer, der kan relateres til CF, inkluderer:
- Røntgen- eller CT-scanning på brystet
- Fækalt fedt test
- Lungefunktionstest
- Måling af bugspytkirtelfunktion (afføring af bugspytkirtel elastase)
- Secretin stimulation test
- Trypsin og chymotrypsin i afføring
- Øvre GI og tyndtarmsserier
- Lungekulturer (opnået ved sputum, bronkoskopi eller halspind)
En tidlig diagnose af CF og behandlingsplan kan forbedre både overlevelse og livskvalitet. Opfølgning og overvågning er meget vigtig. Når det er muligt, skal pleje behandles på en specialklinik for cystisk fibrose. Når børn når voksenalderen, skal de overføre til et specialcenter for cystisk fibrose for voksne.
Behandling af lungeproblemer inkluderer:
- Antibiotika til forebyggelse og behandling af lunge- og bihuleinfektioner. De kan tages gennem munden eller gives i venerne eller ved åndedrætsbehandlinger. Mennesker med CF kan kun tage antibiotika, når det er nødvendigt eller hele tiden. Doser er ofte højere end normalt.
- Inhalerede lægemidler for at hjælpe med at åbne luftvejene.
- Andre lægemidler, der gives ved en åndedrætsbehandling til tynd slim og gør det lettere at hoste, er DNA-enzymbehandling og stærkt koncentrerede saltopløsninger (hypertonisk saltvand).
- Influenza vaccine og pneumokok polysaccharid vaccine (PPV) årligt (spørg din læge).
- Lungetransplantation er en mulighed i nogle tilfælde.
- Iltbehandling kan være nødvendig, da lungesygdommen bliver værre.
Lungeproblemer behandles også med terapier for at fortynde slimet. Dette gør det lettere at hoste slim ud af lungerne.
Disse metoder inkluderer:
- Aktivitet eller motion, der får dig til at trække vejret dybt
- Enheder, der bruges i løbet af dagen til at hjælpe med at rense luftvejene for for meget slim
- Manuel bryst percussion (eller brystfysioterapi), hvor et familiemedlem eller en terapeut let klapper personens bryst, ryg og område under armene
Behandling af tarm- og ernæringsproblemer kan omfatte:
- En særlig diæt med højt proteinindhold og kalorier til ældre børn og voksne
- Bukspyttkjertelenzymer, der hjælper med at absorbere fedt og protein, som tages med hvert måltid
- Vitamintilskud, især vitamin A, D, E og K
- Din udbyder kan rådgive andre behandlinger, hvis du har meget hårde afføring
Ivacaftor, lumacaftor, tezacaftor og elexacaftor er medicin, der behandler visse typer CF.
- De forbedrer funktionen af et af de defekte gener, der forårsager CF.
- Op til 90% af patienterne med CF og berettiget til en eller flere af disse lægemidler alene eller i kombination.
- Som et resultat er der mindre ophobning af tykt slim i lungerne. Andre CF-symptomer forbedres også.
Pleje og overvågning derhjemme bør omfatte:
- Undgå røg, støv, snavs, dampe, husholdningskemikalier, pejsrøg og skimmel eller meldug.
- At give masser af væsker, især til spædbørn og børn i varmt vejr, når der er diarré eller løs afføring eller under ekstra fysisk aktivitet.
- Træner 2 eller 3 gange hver uge. Svømning, jogging og cykling er gode muligheder.
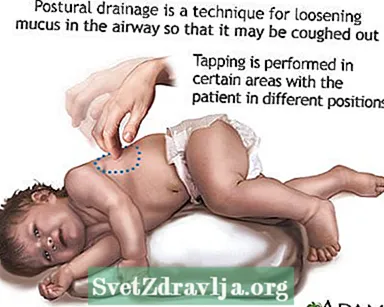
- Rydning eller frembringelse af slim eller sekreter fra luftvejene. Dette skal gøres 1 til 4 gange hver dag. Patienter, familier og plejepersonale skal lære at udføre bryst percussion og postural dræning for at hjælpe med at holde luftvejene fri.
- Ingen kontakt med andre mennesker med CF anbefales, da de kan udveksle infektioner (gælder ikke familiemedlemmer).
Du kan lette sygdomsstresset ved at tilslutte dig en cystisk fibrose-supportgruppe. Deling med andre, der har fælles oplevelser og problemer, kan hjælpe din familie til ikke at føle sig alene.
De fleste børn med CF forbliver ved godt helbred, indtil de når voksenalderen. De er i stand til at deltage i de fleste aktiviteter og gå i skole. Mange unge voksne med CF slutter college eller finder job.
Lungesygdom forværres til sidst til det punkt, hvor personen er handicappet. I dag er den gennemsnitlige levetid for mennesker med CF, der lever til voksenalderen omkring 44 år.
Døden skyldes oftest lungekomplikationer.
Den mest almindelige komplikation er kronisk luftvejsinfektion.
Andre komplikationer inkluderer:
- Tarmproblemer, såsom galdesten, tarmblokering og rektal prolaps
- Hoste op blod
- Kronisk åndedrætssvigt
- Diabetes
- Infertilitet
- Leversygdom eller leversvigt, pancreatitis, galde cirrose
- Underernæring
- Næsepolypper og bihulebetændelse
- Osteoporose og gigt
- Lungebetændelse, der fortsætter med at komme tilbage
- Pneumothorax
- Højre-sidet hjertesvigt (cor pulmonale)
- Kolorektal kræft
Ring til din udbyder, hvis et spædbarn eller barn har symptomer på CF og oplever:
- Feber, øget hoste, ændringer i sputum eller blod i sputum, appetitløshed eller andre tegn på lungebetændelse
- Øget vægttab
- Hyppigere afføring eller afføring, der lugter ildelugtende eller har mere slim
- Hævet mave eller øget oppustethed
Ring til din udbyder, hvis en person med CF udvikler nye symptomer, eller hvis symptomerne bliver værre, især alvorlige vejrtrækningsbesvær eller hoster op med blod.
CF kan ikke forhindres. Screening af dem med en familiehistorie af sygdommen kan detektere CF-genet i mange bærere.
CF
- Enteral ernæring - barn - håndtering af problemer
- Gastrostomi fodringsrør - bolus
- Sådan trækker du vejret, når du er åndenød
- Jejunostomi fodringsrør
- Postural dræning
 Clubbing
Clubbing Postural dræning
Postural dræning Knebede fingre
Knebede fingre Cystisk fibrose
Cystisk fibrose
Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor / ivacaftor hos forsøgspersoner med cystisk fibrose og F508del / F508del-CFTR eller F508del / G551D-CFTR. Am J Respir Crit Care Med. 2018; 197 (2): 214-224. PMID: 28930490 pubmed.ncbi.nlm.nih.gov/28930490/.
Eagan ME, Schechter MS, Voynow JA. Cystisk fibrose. I: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, red. Nelson lærebog om pædiatri. 21. udgave Philadelphia, PA: Elsevier; 2020: kap 432.
Farrell PM, White TB, Ren CL, et al. Diagnose af cystisk fibrose: retningslinjer for konsensus fra Cystic Fibrosis Foundation. J Pediatr. 2017; 181S: S4-S15.e1. PMID: 28129811 pubmed.ncbi.nlm.nih.gov/28129811/.
Graeber SY, Dopfer C, Naehrlich L, et al. Effekter af lumacaftor / ivacaftor-behandling på CFTR-funktion hos Phe508del homozygote patienter med cystisk fibrose. Am J Respir Crit Care Med. 2018; 197 (11): 1433-1442. PMID: 29327948 pubmed.ncbi.nlm.nih.gov/29327948/.
Grasemann H. Cystisk fibrose. I: Goldman L, Schafer AI, red. Goldman-Cecil medicin. 26. udgave Philadelphia, PA: Elsevier; 2020: kap 83.
Rowe SM, Hoover W, Solomon GM, Sorscher EJ. Cystisk fibrose. I: Broaddus VC, Mason RJ, Ernst JD, et al., Red. Murray og Nadels lærebog om respiratorisk medicin. 6. udgave Philadelphia, PA: Elsevier Saunders; 2016: kapitel 47.
Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor hos patienter med cystisk fibrose homozygot for phe508del. N Engl J Med. 2017; 377 (21): 2013-2023. PMID: 29099344 pubmed.ncbi.nlm.nih.gov/29099344/.
Se

Selena Gomez gik til boksning til sin første træning efter nyretransplantation